Årsaken til Huntingtons Sykdom
-
Om Huntingtons sykdom
- Årsaken til Huntingtons Sykdom
- Symptomer
- Fasene i Huntingtons sykdom
- Finnes det en kur?
-
Erfaringer, Huntington sykdom (historier/artikler)
- Tryggere og mer næringsrike måltider
- Avdeling for HS-pasienter i Oslo
- Fjerne skam med økt kunnskap
- Livet med den sjeldne sykdommen
- Nasjonal ressurs, psykiater Elena Selvåg
- Slik kan sykepleiere gi bedre pleie til døende pasienter med Huntingtons sykdom
- Presidentbesøk for ressurssenter
- Å gjere kvarandre gode
- Mai måned med fokus på HS
HTT-genet:
HS skyldes en sykdomsfremkallende feil i genet HTT (Huntingtin-genet), tidligere også kalt IT15. Arvestoffet vårt finnes inne i kjernen til alle kroppens celler som lange tråder med DNA, og kalles kromosomer. På DNA-tråden finnes oppskrifter for hvordan cellen skal bygge proteiner, som en kodet sekvens av fire forskjellige baser, og benevnes med bokstavene A, G, C og T. Proteiner er store og komplekse molekyler som utfører de fleste spesialiserte oppgavene i cellen. En slik oppskrift som koder for et bestemt protein, kalles et gen.
I 1993 påviste forskere at årsaken til HS er en bestemt endring i HTT, som sitter på kromosom 4 og koder for proteinet huntingtin. I en del av oppskriften forekommer mange repetisjoner av sekvensen C-A-G, som koder for aminosyren glutamin, som er en byggestein i proteinet.
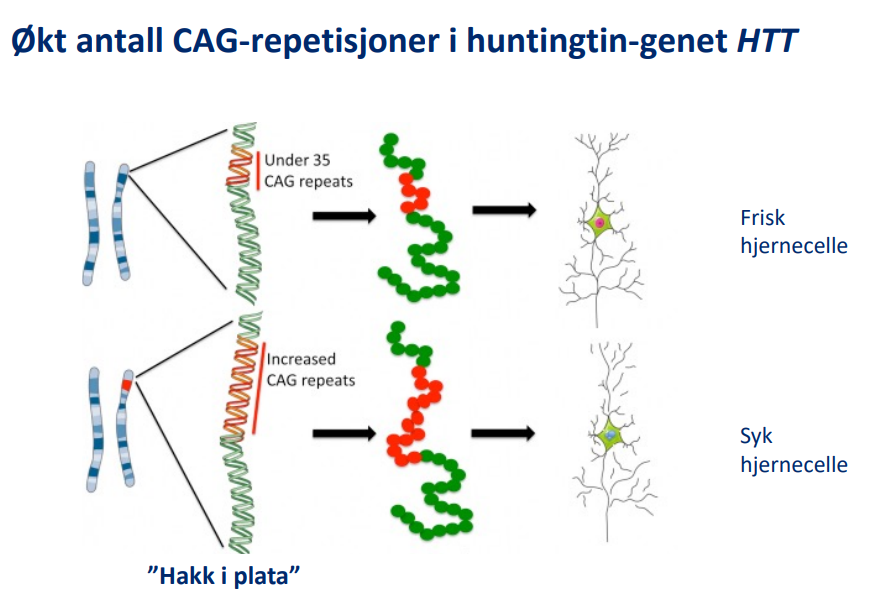
(Bildet er fritt oversatt, fra eurostemcell.org)
-Det har vært vanlig å si at sykdommen kan bryte ut dersom man har 36 eller flere repetisjoner. Mutasjonen fører til at det blir et unormalt antall repetisjoner av en av byggeklossene, en aminosyre som kalles glutamin, og huntingtinet får endrede egenskaper og skader nervecellene. Antallet CAG-repetisjoner kan endre seg til neste generasjon. Det er derfor mulig at en person kan rammes av HS selv om sykdommen ikke har forekommet i slekten tidligere. Med bakgrunn i forskning i andre land anslår vi at det kan oppstå 1-2 tilfeller av «nyoppstått» HS hvert år i Norge.
• Opptil 26 repetisjoner regnes som normalt.
• Personer som har 40 eller flere slike CAG-repetisjoner i ett av sine HTT-gener, vil i løpet av livet utvikle HS.
• Mellom 36 og 39 repetisjoner er det en gråsone der sykdommen vil bryte ut hos noen, men ikke hos alle.
Hvor skjer skaden?
HS rammer alle celler i kroppen, men det er særlig skaden i hjernen som gir symptomer. Forandringene i hjernen skjer sakte og ses først i et område sentralt i hjernen som kalles «striatum». Hjernen har en viss reservekapasitet, men når denne er brukt opp vil den som er rammet av HS utvikle symptomer. Man går da fra å være presymptomatisk genbærer til å ha kliniske tegn og symptomer på HS.
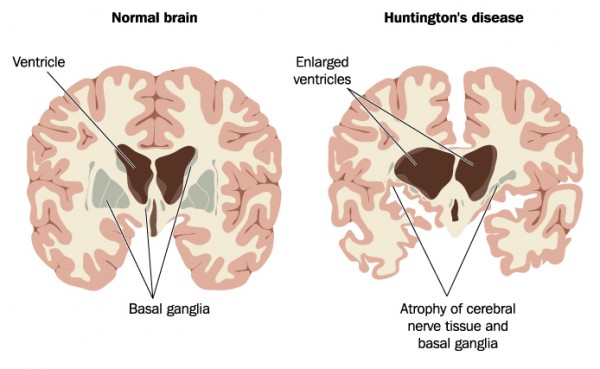
(PLAMB / SHUTTERSTOCK)
Er det sammenheng mellom gener og når man blir syk?
HS rammer vanligvis i voksen alder, og symptomene starter som regel når man er mellom 30 og 55 år. Noen ytterst få får HS i ung alder, før fylte 20 år, og da snakker man om «juvenil HS». I undersøkelser av et stort antall personer med HS har man påvist at antall CAG-repetisjoner i noen grad er knyttet til alder for når man utvikler symptomer på HS. Hvis man har et høyt antall CAG-repetisjoner er det større sjanse for at sykdommen rammer i yngre alder, og typisk har de som får sykdommen i høy alder et lavt antall CAG-tall. Det er likevel stor variasjon fra individ til individ, og antall CAG-repetisjoner kan derfor ikke brukes til å forutsi sikkert når en person vil få sykdommen. Nyere forskning har dessuten påvist at andre gener enn HTT-genet kan ha stor påvirkning på når sykdommen utvikler seg. Vi snakker her sannsynligvis om gener som påvirker cellenes evne til å håndtere de skadelige effektene av det endrede huntingtinet.
Hvor vanlig er HS?
Forekomsten av HS på verdensbasis er et sted mellom 5-10 personer per 100.000. Anslagsvis er det 350 personer med symptomer på HS i Norge. I tillegg vil det anslagsvis være 700 personer som er presymptomatiske bærere av arveanlegget for HS, og som ennå ikke har utviklet symptomer på sykdommen. HS forekommer i alle etniske grupper, over hele verden.
Kilder hentet fra ; oslo-universitetssykehus.no/veileder/huntingtons-sykdom